510(k) Exempt Medical Device Guide
Navigating the intricacies of medical device class regulations is crucial for manufacturers and innovators. In this article, we delve into what it means to be 510(k) exempt, shedding light on its significance, benefits, and considerations. We hope to help you streamline your regulatory path and bring innovative medical solutions to the market more efficiently!
What is a 510(k)?
A 510(k) is a premarket submission made to the Food and Drug Administration (FDA) to demonstrate that the device being marketed is safe and effective. The purpose of the submission is to demonstrate that your medical device is substantially equivalent to an existing device (predicate device) on market today. A 510(k) is required if the device was manufactured to the inventor’s own specifications and is intended to be marketed in the US. FDA clearance through the 510(k) process means the agency agrees with the manufacturer that the medical device is similar to a previously approved predicate device.
What does it mean to be 510(k) exempt?
Some medical devices are not required to undergo the traditional 510(k) submission process to obtain clearance from the FDA before marketing in the US. Devices that are considered low risk typically have well established safety profiles and do not present significant risks to patients; these devices may qualify for exemption. If the device is exempt from submitting a 510(k), it still needs to comply with applicable regulatory requirements in accordance with FDA guidelines, such as medical device labeling, good manufacturing processes (GMPs), quality systems, and post-market surveillance. Any change to the device design or intended use may require a new submission to ensure continued compliance.
Who is required to submit a 510(k) for medical devices?
There are four groups indicated by the FD&C Act and 510(k) regulation (21 CFR 807) who would be responsible for the completion of a 510(k) submission for medical devices. They include:
- Domestic manufacturers introducing a device to the U.S. market
- Specification developers introducing device to the U.S market
- Repackers or relabelers in a medical device supply chain
- Foreign manufacturers and their representatives introducing a device to the U.S. market.
What is required for a 510(k)?
To qualify for a 510(k), submitters must compare their device to one or more legally marketed predicate devices and make and support their substantial equivalence claims. A device is substantially equivalent if, in comparison to the predicate if it:
- has the same intended use; and same technical characteristics; OR
- has the same intended use; and different technological characteristics; AND
- does not raise different questions of safety and effectiveness; AND
- the information submitted to FDA demonstrates that the device is as safe and effective as the legally marketed device.
510(k) application submissions require specific information and documentation, as well as appropriate methods, to demonstrate the substantial equivalence of a new medical device to a legally marketed predicate device. In contrast, PMA (Premarket Approval) is required for high-risk Class III medical devices that do not have a predicate device. The specific requirements for a 510(k) submission can differ based on the device type, complexity, and risk classification.
The following are key deliverables typically required within a 510(k) submission for medical devices:
- Cover letter
- Device description
- Indications for use
- Substantial equivalence discussion (predicate device comparison)
- Performance testing data (including clinical and non-clinical studies)
- Biocompatibility
- Software and firmware
- Manufacturing information (including declaration of conformity)
- Sterilization and packaging (including validation of device’s sterility and integrity)
- Labeling
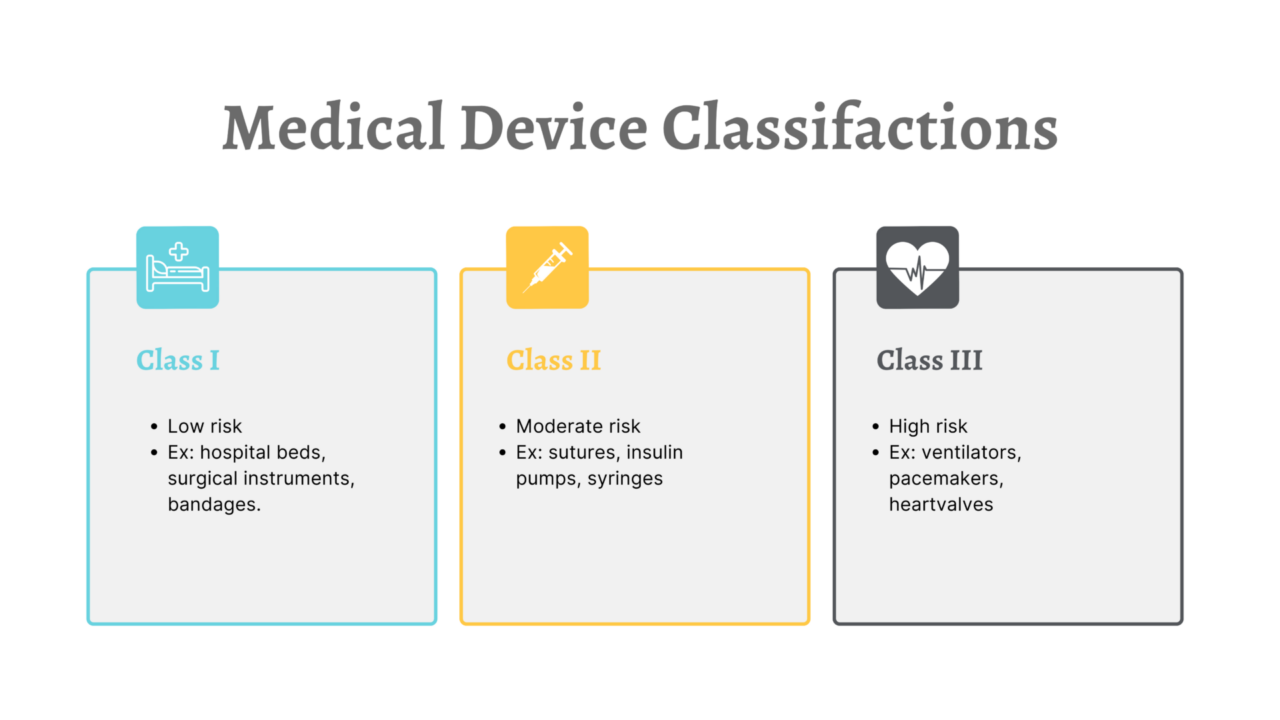
Most class I and class II devices are exempt from 510(k) requirements. Devices that may be 510(k) exempt include:
- Preamendment devices (devices legally marketed in the US prior to May 28, 1976)
- Class I devices specifically exempted by FDA or classified as class I under section 513 (with certain exceptions)
- Examples of Class I devices: manual surgical instruments, examination gloves, tongue depressors, non-powered orthopedic devices.
- Class II devices with special controls
- Examples of Class II devices: powered wheelchairs, dental floss, pregnancy test kits.
- Class II devices with published performance standards
- Examples of Class III devices: dental hand instruments, intraocular lenses, in-clinic electronic thermometers.
- Certain Class II devices with Premarket Notification exemption
A list of class I and class II devices exempt from 510(k) requirements is available on the FDA website.
How Gilero can help you with your medical device 510(k) submission
If your device qualifies for a 510(k) submission, our experienced regulatory team can help you from any point in the approval process. Gilero can provide on-going consultation services throughout a product’s life cycle, or as new requirements are published by regulatory agencies. Our regulatory experts at Gilero can perform regulatory assessments, select predicate device(s), as well as author and submit the 510(k) on your behalf. After submission, we participate and manage our client’s involvement in the FDA’s review process, including facilitating any FDA inspections. Gilero will work with the client to satisfy any requests and requirements resulting from the review process, resulting in a more efficient path to market.
Ready to turn your idea for a medical or ocular drug delivery device into a reality?
Talk with an expert today.