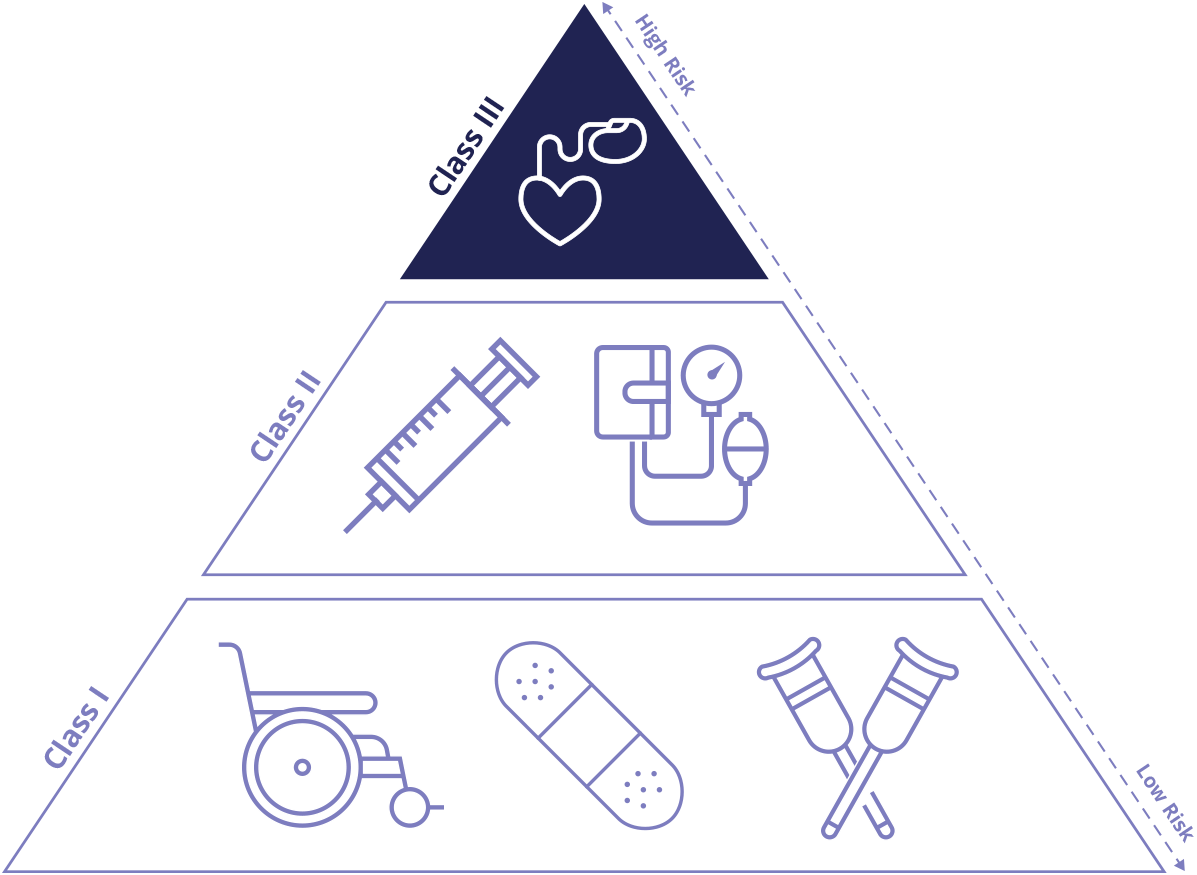
What is Medical Device Classification
All medical devices in the United States go through a review process by the US Food and Drug Administration (FDA) before they can be marketed for patient use. High risk devices such as those implanted into the body are more regulated than those with minimal risk, such as a pair of tweezers or bandages. Therefore, the FDA assigns every medical device a “class” based on its risk profile, which is the potential threat posed to the safety of the patient if something were to go wrong. The three classifications include: Class I, II, and III. Each class is based on the level of control necessary to assure the safety and effectiveness of the device.
Types of Medical Device Classifications
Class I Medical Devices:
Lowest risk medical devices, these devices present minimal potential harm to the user. Examples include bandages, and handheld surgical instruments. Around 47% of medical devices fall into this category.
Class II Medical Devices:
Intermediate risk medical devices, examples include CT scanners, catheters, and infusion pumps. 43% of medical devices are Class II.
Class III Medical Devices:
High risk devices, these devices are especially important to health or sustaining life. Examples include pacemakers, orthopedic implants, and artificial valves. About 10% of medical devices are Class III.

Why is Medical Device Classification Important?
A key reason for classifying medical devices is to provide reasonable assurance of their safety and effectiveness for the user. Product classification determines the regulatory rules and requirements you must comply with before you can sell your product. Each class has their own set of regulations, and knowing the class your device falls into is critical to prepare the necessary documentation for FDA approval. The type of classification of your device also is an important component of how much it will cost and the time it will take to bring your product to market.
How to Classify your Medical Device
The first step in classifying your device includes establishing the intended use and indications for your medical device. Intended use refers to the general purpose of your device. The device’s indications include the disease or condition that the device is meant to prevent, treat, or diagnose.
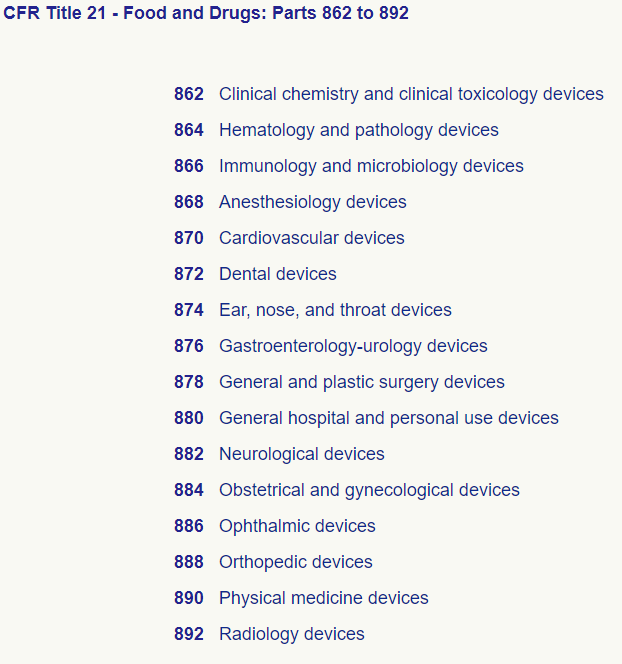
The second step includes specifying the panel that aligns with your device out of the 16 different medical device specialties grouped by the FDA. These panels are listed in CFR Title 21: Parts 862 to 892.
The third and final step includes finding the FDA product codes that are applicable to your device by searching in the FDA Product Classification Database. The search is conducted through entering the regulation numbers selected from the second step. A detailed listing of information will populate that includes a product code and corresponding device class.
What types of medical devices qualify for 510(k) exemptions?
The class to which your device is assigned determines the type of premarketing submission/application required for FDA clearance to market.
Most Class I and Class II devices are exempt from 510(k) requirements. Devices that may be 510(k) exempt include:
- Preamendment devices
- Class I devices specifically exempted by FDA or classified as class I under section 513 (with certain exceptions)
- Class II devices specifically exempted by the FDA
A listing of Class I and Class II devices exempt from 510(k) requirements is available on the FDA website.
For Class III devices, a premarket approval application (PMA) will be required unless your device is a preamendments device (on the market prior to the passage of the medical device amendments in 1976, or substantially equivalent to such a device). In that case, a 510k will be the route to market.
Medical Device Classification with Gilero
Gilero’s in-house regulatory experts provide guidance and recommendations for classifying your device while considering the intended use, technology, regulatory landscape, and commercialization strategy. With this assessment in hand, the output of our design and development process provides a complete package for regulatory submissions and transfer into commercial production. Along with preparation and submission, Gilero can provide ongoing support for your product throughout every phase of its life cycle.
Gilero follows FDA 21 CFR Part 820 as part of our Quality Management System (QMS), which ensures that all medical devices created and developed within the US market follow satisfactory quality processes at all stages of development. In addition, Gilero is ISO 13485 certified, which ensures that your medical device is safe and consistent when it comes to its design, development, and manufacturing processes.
